
Fejlett terápiás gyógyszerkészítmény
|
|
Ezt a szócikket némileg át kellene dolgozni a wiki jelölőnyelv szabályainak figyelembevételével, hogy megfeleljen a Wikipédia alapvető stilisztikai és formai követelményeinek. |
|
|
Ezt a szócikket tartalmilag és formailag is át kellene dolgozni, hogy megfelelő minőségű legyen. További részleteket a cikk vitalapján találhatsz. Ha nincs indoklás a vitalapon, bátran távolítsd el a sablont! |
A fejlett terápiás gyógyszerkészítmények (angol: advanced therapy medicinal product; röv.: ATMP) az innovatív gyógyszerek széles körét képviselik, amelyek a betegségek kezelésére szolgáló sejt- és génalapú eljárásokat alkalmazzák.[2] Ez az elnevezés a sejt-, szövet- és génterápiás gyógyszerkészítmények elnevezését jelenti. Az ATMP-k különböznek a hagyományos kis molekulájú gyógyszerektől (kis molekulatömegű kémia vegyületek) és a biológiai gyógyszerektől (nagy molekulatömegű proteinek), mivel aktív sejteket vagy genetikai konstrukciókat tartalmaznak, amelyek metabolikus, immunológiai, genetikai vagy egyéb nem farmakológiai hatásmechanizmussal gyógyítanak.[3] Az ATMP-k technikailag rendkívül igényesek a tervezésben és gyártásban is, viszont napjainkban nagyon korlátozott kereskedelmi sikereket értek el.
Két évtizedes kudarcai után napjaink egy izgalmas idő a sejt- és génterápiák terén, mivel úttörő előrehaladást mutatunk számos területen a vérrák, az immunhiányos betegségek és a szemészet területén. Az ágazat gyorsan fejlődik, hogy megfeleljen azoknak a kihívásoknak, amelyek hathatós új terápiás eljárásokat képesek létrehozni.

A CAR (kiméra antigén receptor) T-sejtes terápiák példájánál a betegek saját T-sejtjeit vérből izolálják és genetikailag újraprogramozzák, hogy felismerjék és megöljék a tumorsejteket, mielőtt újra bejuttatják a beteg véráramába. A betegek sejtjeinek a szakma szabályai szerinti környezetben való megbízható és biztonságos módosítása és a megfelelő sejtek kezelése a betegek számára, valamint a betegek nyomon követése és e láncolata felügyelet kényes és költséges folyamat. A jelenleg fejlesztés alatt álló legtöbb sejt- és génterápia ritka betegségeket céloz meg, a magasabb árat ezzel lehet indokolni a gyártókhoz való megtérülés biztosítása érdekében, ami az innováció előmozdításához és a hatékonyabb gyógyszerek kifejlesztéséhez szükséges feltétel. A CAR-T termékekre vonatkozóan három amerikai piaci engedély már megjelent, és a további termékek széles választéka várható.[4]
A fejlett terápiáknak valóban új paradigmára van szükségük – különösen a kereskedelmi modellek esetében. Míg a sejt- és génterápiák potenciális paradigmaváltást jelentenek a rák és a ritka genetikai rendellenességek kezelésére, ezeknek a nem hagyományos terápiáknak a finanszírozása és megtérítése nagy kihívást jelent.[5] A mai napig a gyártók, a tudósok és a klinikusok arra törekednek, hogy lehetővé tegyék e személyre szabott gyógyszerek kifejlesztését és a rák összetett jellegének kezelését minden egyes beteg számára, és most a gyártók a hangsúlyt arra irányítják, hogy ezeket az innovatív terápiákat piacra dobják. A kérdés az, hogyan tudják a gyártók termékeiket navigálni a komplex egészségügyi ökoszisztémában úgy, hogy azokat az innovatív gyógyszereket kapják a betegek, amire szükségük van.
- A hagyományos biológiai ismeretek előrehaladása és a terápiás beavatkozások komplexitásának növekedése:[6]
| Kismolekulájú modulátorok | Protein augmentáció | Antitestek | Peptidek és nukleinsavak | Génkorrekció és augmentáció | Sejtterápiák és regeneratív orvoslás |
|---|---|---|---|---|---|
|
|
|
|
|
|
A fejlett terápiás gyógyszerkészítmények (ATMP) fejlesztésének rövid története[szerkesztés]

Az ATMP nevét az Európai Bizottság 1394/2007 rendelete vezette be, bár a sejteket vagy géneket alkalmazó gyógyszerek már évek óta fejlesztés alatt álltak. Ezeknek a termékeknek a fejlesztése azonban első alkalommal Thomson és munkatársai (1998) által vezetett egyetemi klinikán folyt,[7] amely a molekuláris szerkesztési és szövetkultúra technikák terén elért tudományos eredményeken alapult.
A szabályozási környezetet az embrionális őssejtek megjelenése és az in vitro manipulációs potenciáljuk a figyelmet erősen ráirányította az ATMP-re (Thomson és mások, 1998). Az embrió eredetéhez kapcsolódó bioetikai kérdések és az elkülönített embriók létrehozásához használt in vitro megtermékenyítési módszerek is (Wainwright és mások, 2006)[8] számos aggályt vetettek fel. Az alkalmazott technológiák gyors fejlődésére adott válaszként az Európai Parlament elfogadta az 1394/2007 rendeletet azzal a feltétellel, hogy 2008-ban hatályba lép, és a következő hároméves átmeneti időszakot engedélyezik a már regisztrált termékek nyilvántartásba vételéhez a nemzeti piacon.
A központosított piacfelhatalmazási folyamathoz történő megfeleléshez szükséges idő becslése rendkívül optimista volt, de ennek ellenére 2017-ig még mindig csak hat engedélyezett ATMP van forgalomban, amelyek közül három termék kereskedelmi okok miatt kivonult a piacról (PEI, 2016).[9] Az USA-ban az FDA Sejt, Szövet- és Génterápiák Hivatala engedélyeket adott ki hét olyan termékre, amelyek hasonlóak az európai ATMP-khez (FDA, 2017).
A kritikus és igényes fejlesztés az ATMP területen, mint általában, új technológiákkal jelenik meg. Az új immunológiai terápiák megjelenése a kiméra antigén receptor T- sejt génszerkesztő technika alkalmazása rendkívüli esemény volt (Fellmann et al., 2017). Ezek az új termékek egyértelmű piaci potenciált mutattak a kiemelkedő klinikai eredmények révén, amelyek könnyebben érthetők a szabályozási folyamatban és világos cselekvési módban. Tény, hogy a CAR-T-sejtek a gyógyszer-konjugált antitestek evolúciójának tekinthetők, vagyis egy olyan gyógyszer, amely élő sejt is. Mindazonáltal az új termékek hosszú távú életképességét értékelni kell, mivel az alkalmazott üzleti modellt nem vizsgálják a gyártás és a megtérülés tekintetében.
- Kontrollált alkalmazások száma[10]
2009 2010 2011 2012 2013 2014 2015 2016 Szomatikus sejtterápiás termék (SCTMP) 5 6 3 3 5 5 11 2 Génterápiás termék 3 6 3 3 0 5 3 8 Szövet módosított termék 1 9 4 5 5 11 12 30 Nem ATMP 2 1 1 2 3 1 1 1
- Az EMA által az ATMP-k osztályozására vonatkozó útmutatások száma. A fejlesztés alatt álló termékek számának képviselője az egyes besorolásokon belül.
Fejlett terápiás gyógyszerek kategóriái[szerkesztés]

Az Európai Gyógyszerügynökség (EMA) négy fő kategóriába sorolja az ATMP-ket:[11]
- szöveti biomérnöki úton készült gyógyszerkészítmények (TEP)
- szomatikus sejtterápiás gyógyszerek (SCTMP)
- génterápiás gyógyszerek (GTMP)
- kombinált fejlett terápiás gyógyszerkészítmény kATMP.
A TEP-k általában regeneratív eljárások, amelyek magukban foglalják az őssejtek alkalmazását a károsodott szövetek, például a szív, a porc, a csont vagy az idegszövet-javítás hosszú távú regenerálására és / vagy cseréje. Az SCTMP-k olyan sejteket foglalnak magukban, amelyeket úgy terveztek, hogy más célt szolgáljanak eredeti funkciójukban, gyakran rövid távú intervencionista hatást gyakorolva a beteg élettanára a betegség leküzdésére (például onkológiai indikációk).
A GTMP-k nem tartalmaznak aktív sejteket, hanem genetikai mérnöki eszközöket használnak a pácienssejtek (egy részhalmaza) genetikai összetételének módosítására aktív rekombináns nukleinsavakon keresztül. A kombinációs termékek olyan gyógyászati eszközt képeznek, amely egy aktív sejt, pl. ahol az élő sejtek a mesterséges kapszulába vannak beágyazva.
A sejtterápiák autológok vagy allogének lehetnek. Az autológ terápiák a kezelt betegből származnak, és egy újrahasznosítás előtt meghatározott protokollon keresztül gyártják. Az autológ terápiák az immunológiai kompatibilitás előnyeit kínálják, de általában összetettebb ellátási láncot igényelnek. Az allogén terápiák donorsejtekből származnak, és a mester- és munkaképcellák felépítése nagymértékben történik, amely lehetővé teszi az off-the-shelf eloszlását és alkalmazását.[12]
- A legfontosabb meghatározások jogi megfogalmazása a 2.1 cikkelynek megfelelően az 1394/2007 / EK rendelet szerint:
Tárgy Jogi meghatározás szövege Fejlett terápiás gyógyszerkészítmény (ATMP) - a) a 2001/83 / EK irányelv I. mellékletének IV. részében meghatározott génterápiás gyógyszerkészítmény,
- b) a 2001/83 / EK irányelv I. mellékletének IV. részében meghatározott szomatikus sejtterápiás gyógyszerkészítmény,
- c) az 1394/2007 / EK rendelet 2. cikke (1) bekezdésének b) pontjában meghatározott, módosított szöveti termék
Szöveti biomérnöki úton készült gyógyszerkészítmények - a) mérnöki sejteket vagy szöveteket tartalmaz vagy azokból áll, és
- b) olyan humán szövetek regenerálódására, javítására vagy helyettesítésére, amelyek emberi tulajdonságokkal rendelkeznek, vagy amelyeket emberekben alkalmaznak vagy adnak be.
Szomatikus sejtterápiás gyógyszerek (SCTMP) - a) Olyan sejteket vagy szöveteket tartalmaz vagy azokból áll, amelyek jelentős manipulációnak vannak alávetve annak érdekében, hogy a tervezett klinikai alkalmazás szempontjából releváns biológiai jellemzőket, élettani funkciókat vagy szerkezeti tulajdonságokat megváltoztatták, vagy azokat a sejteket vagy szöveteket, amelyeket nem szándékoznak használni ugyanazon lényeges funkció (k) a recipiensben és a donorban;
- b) olyan tulajdonságokkal rendelkeznek, amelyek emberi betegségek kezelésére, megelőzésére vagy diagnosztizálására alkalmasak, vagy amelyeket emberi szervezetben alkalmaznak vagy adnak be, annak érdekében, hogy sejtjei vagy szövetei farmakológiai, immunológiai vagy metabolikus hatása révén legyenek.
Génterápiás gyógyszerek (GTMP) - a) olyan hatóanyagot tartalmaz, amely egy genetikai szekvencia szabályozására, javítására, helyettesítésére, hozzáadására vagy törlésére szolgáló, vagy embereknek beadott rekombináns nukleinsavból áll;
- b) terápiás, profilaktikus vagy diagnosztikai hatása közvetlenül a rekombináns nukleinsav-szekvenciájához vagy a szekvencia genetikai expressziójának termékéhez kapcsolódik.
Kombinált fejlett terápiás gyógyszerkészítmény (kombinált ATMP) - a) a termék szerves részeként a 93/42 / EGK irányelv 1. cikke (2) bekezdésének a) pontja vagy egy vagy több aktív beültethető orvostechnikai eszköz A 90/385 / EGK irányelv 1. Cikke (2) bekezdésének c) pontja, és
- b) sejtes vagy szövetrészének életképes sejteket vagy szöveteket kell tartalmaznia, vagy c) a nem életképes sejteket vagy szöveteket tartalmazó sejtes vagy szöveti részének alkalmasnak kell lennie arra, hogy az emberi testre olyan hatással járjon, amely elsődlegesnek tekinthető a eszközöket.
Jelentősebb terápiás szektorok[szerkesztés]



Az 1980-as években és a 2000-es évek elején bekövetkezett kezdeti aggodalmak és csalódást keltő tanulmányok után a sejt- és génterápiák az orvostudomány élvonalában vannak, és számos klinikai vizsgálatban példátlan hatékonyságot mutattak ki. Amint egyre több kísérlet fordul elő, a betegek sikertörténetei egyre nőnek, és egyre több gyártó fektet be vagy partnerként fejleszt tovább a következő divathullámig a gyógyszerkutatásba. 2015-ben a Journal of Immunology Research szerint közel 100 klinikai vizsgálatot végeztek CAR (kiméra antigén receptor) T-sejtes terápiákkal kapcsolatban. Az immunonkológia jelenleg a fejlett terápiás szektor uralkodó terápiás csoportja, amely a 2016-os év összes klinikai vizsgálatának mintegy felét teszi ki.[14] A CAR-T a legfontosabb hajtóereje ennek, és a CAR-T piac várhatóan 8,5 milliárd dollárra becsülhető 2028-ra.[15] Az ágazat növekedése nagyrészt a genetika onkológiában betöltött szerepének kifejlődésével, a lentivirális vektorbiztonság és az élvonalbeli genetikai mérnöki eszközök, például a CRISPR/ Cas9 érvényesítésével összhangban képes volt vezető kutatási területté előlépni. A nem-specifikus sejtabláció paradigmája, amely az immunrendszer elpusztításával és más káros mellékhatásokkal jár, a hagyományos kemoterápiás és radioterápiás kezelés paradigmája, az immunonkológiai megközelítések célja a természetes immunválasz kiaknázása és növelése a korrekt rákkutatásban és gyógyításban.
Számos eljárás létezik arra, hogy a celluláris terápiák alkalmazhatóak az onkológiában: az első nagy sejtalapú eljárás a hemopoetikus őssejt-transzplantáció a kemoterápiát és / vagy sugárkezelést követően, de konkrétabb terápiás eljárás is bevezetésre került, mint például – beleértve a dendritikus sejtvakcinákat – a szerkesztett T-sejt-receptorok (TCR) és kiméra antigén receptor T-sejtek. A CAR-T-k magában foglalják a T-sejt-receptorok genetikai konstrukcióját specifikus tumorantigénekre kifejlesztve, ami T-sejteket eredményez, amelyek mind közvetlenül támadják a tumorsejteket, és szélesebb immunválaszt indítanak be. A CAR-T-k napjainkban a legfontosabb technológiatípus az ATMP térben, több mint 100 klinikai kísérlet van folyamatban 2016-ban, a 2015-ös 250%-os emelkedés és közel 600 millió dollár kockázati tőkét jelent.[16]
Ugyanígy a nyilvános piac tükrözte ezt az érdeklődést, a hat vezető fejlett terápiás céggel (amelyek többsége CAR-T termékeket fejlesztett ki), közel 1 milliárd dollárt emeltek IPO-jukat. A CAR-T-k legfontosabb indikációs kategóriája a malignus limfómák, ami a világ összes rosszindulatú betegségének 3,37%-át teszi ki. Az USA-ban 2017-ben várhatóan 174 000 leukémia, limfóma, nem-Hodgkin-lymphoma és myeloma a vér daganatos megbetegedéseiben a CAR-T termékek által gyógyítható. A szolid tumorokban korlátozott és vegyes adatokkal aggodalomra ad okot, hogy a CAR-T-k hamarosan meglehetősen korlátozott piacot ölel fel. Számos vállalat felismerte ezt, és további CAR-T-ket fejleszt ki az alternatív sejtek ellen felszíni markerekkel bővíteni potenciális piacát.
A múltbeli és az aktuális betegségekről szóló klinikai vizsgálatok (2010. december 31-i állapot).[17]
Klinikai vizsgálat Vizsgálatok aránya (%) Onkológia 40 Kardiológia 12 Központi idegrendszer 8 Csont- és izomrendszer 7 Fertőzőbetegségek 5 Bőrgyógyászat 5 Endokrin, metabolikus 5 Immunológia 5 Szemészet 4 Hematológia 3 Gasztroenterológia 2 Egyéb 4
Az onkológiában a CAR-T termékek dominálnak a területen, a regeneratív szomatikus sejtterápiákkal, amelyek a kísérletek nagy részét alkotják.
Nem-immunonkológia javallatok[szerkesztés]
Fejlett terápiák fejlesztés alatt állnak az onkológián kívül további immunológiai szempontból releváns indikációkban, ideértve a graft versus host betegséget (GvHD), a cukorbetegség és más autoimmun indikációkat. A főbb sejttípusok a mesenchymális stromális sejtek (MSC-k: MSC egy általánosan meghatározott sejtek heterogén populációja)) és a T-szabályozó sejtek.
Mesenchymális stromális sejtek[szerkesztés]
A MSC-k széles körben bizonyítottan immunmoduláló hatásúak, általában úgy értik, hogy működésüket parakrin mechanizmusok határozzák meg, és jelenleg több mint 250 klinikai vizsgálaton mennek keresztül.[18] A széleskörű korai stádiumú klinikai vizsgálatok ellenére számos vezető szerepet tölt be a területen továbbra is szkeptikusak a valódi hatékonyságuk miatt, a felsorolt kísérletek mindössze 7%-a van a 3. fázisban. A 3. fázisú vizsgálatok közé tartozik a GvHD, a stroke és más szív- és érrendszeri betegségek, gerincvelő javítása, Crohn-betegség, csontbetegségek és agyi paralízis. Az MSC-kben a kereskedelmi érdeklődés jelenleg korlátozott a megbízható klinikai adatok hiánya miatt.
Citotoxikus T-sejtek[szerkesztés]
A citotoxikus T-sejtek az onkológián kívül alkalmazásokkal rendelkeznek, elsősorban a hematopoetikus őssejt-transzplantációt (HSCT) követő vírusfertőzés csökkentésében. Az allogén HSCT-ben szenvedő betegek 20-35%-a cytomegalovírus (CMV) fertőzést termel.[19] A CMV vagy adenovírus (ADV) -specifikus CD8 + T-sejtek támogatása bizonyítottan csökkenti a fertőzés kockázatát, és számos vállalat fejlődik allogén termékeket (pl. Cell Medica, London, Egyesült Királyság).
Szabályozó T-sejtek (T-reg)[szerkesztés]
A szabályozó T-sejtek (T-reg) az immunrendszer hatékony szupresszorai, amelyek endogén módon működnek az immunológiai homeosztázis fenntartásában. A T-regs ellensúlyozza a citotoxikus T-sejtek stimuláló jellegét, fenntartja az önantigénekkel szembeni toleranciát és megakadályozza az autoimmun betegségeket egészséges egyénekben. Kb. 60 klinikai vizsgálat jelenleg folyamatban van, mind a korai tesztelési szakaszban.18 Annak ellenére, hogy a klinikai adatok viszonylagos hiánya ellenére a T-regok széles körben elvárják, hogy az elkövetkező években kereskedelmi fejlődésre számítson.[20]
Sejt- és szövetterápiák, regeneratív orvoslás[szerkesztés]

A sejtalapú fejlett terápiák jelentős hányada "regenerálódási" kezelési módot alkalmaz, és lazán definiálható mint módosított vagy regeneratív gyógyszerek. Az ilyen termékek általában a TEP EMA definíciója alá tartoznak, ellentétben azokkal az SCTMP-kkel, amelyek hajlamosak átmeneti jelleggel járni, és nem feltétlenül jelentenek hosszú távú szöveti javulást. A TERM termékek gyakran tartalmaznak progenitor sejteket. Például HeartCel a szívműtétre (Cell Therapy Ltd), a CTX a stroke-ra és a kritikus végtagi ischaemia-ra (ReNeuron), a MACI a porc-javításra (Vericel) és számos, az égésekre vagy diabéteszes bőr fekélyeire (pl. Dermagraft, Epicel, AmnioExcel). Az EMA iránymutatásai 2016-ban az ATMP-besorolásról a TEP-k számának jelentős növekedését mutatják az előző évhez képest.
Génterápiák[szerkesztés]

A génterápiák a kedvező pénzügyi, szabályozási és piaci ösztönzők miatt korábban a ritka betegségekre utaltak. A ritka betegségekre vonatkozó jogállás lehetővé teszi a magasabb megtéríthető árazási pontokat, amelyek igazolhatják a magasabb fejlesztési költségeket, akár 12 éves piaci kizárólagossági jogokkal kiegészítve. A génterápiákkal foglalkozó vállalatok egyre inkább arra törekszenek, hogy nagyobb és versenyképesebb piacokra lépjenek, amikor a termékfejlesztési infrastruktúrák elég érettek ehhez. Az A és a B hemofília gyógyszer jelöltjei jelenleg nagy jelentőségű génterápiás termékek kategóriájába tartoznak, amelyek számos késői klinikai stádiumú vállalatok között nagy versenyben állnak. A Spark Therapeutics, a Pfizer, a Bayer, a Sangamo, a Freeline Therapeutics, az UniQure és a Shire mind fejlesztenek a hemofília génterápiás termékeit. A génterápiás piaci terekben működő egyéb cégek közé tartozik a Nightstar, a CRISPR Therapeutics, az Editas, a Bluebird, a Celgene, az Intellia, a Pfizer és a Precision Biosciences.
- Az EU-ban forgalomba hozatali engedélyt kapott (AMTP) 2016-ig:[21]
Gyári név Hatóanyag Hatóanyag mennyiség AMTP osztály Jelenlegi jóváhagyás állapota Javallat Ár (€, $) Zalmoxis (MolMed S.p.A.)[22] Az ember kis affinitású idegi növekedési faktor receptor csonkított alakja (ΔLNGFR) és a Herpes simplex-I vírus timidinkinázt (HSV TK Mut2) kódoló retrovirus eredetű vektorral genetikailag modifikált allogén T-sejtek. Zalmoxis 5-20×106 sejt/ml diszperziós infúzió GTMP (ex vivo) Visszavonva (2019-10-11) Kiegészítő terápiaként javasolt haploidentikus vérképzőszervi őssejtek átültetése (HSCT) során olyan felnőtt betegeknél, akiknél magas a vérképzőszervi daganatok kockázata Strimvelis (GlaxoSmithKline Trading Services Limited)[23] Autológ, CD34+ -gyel dúsított sejtfrakció, amely emberi vérképzőszervi ős/progenitor (CD34+) sejtekből származó, emberi adenozin-deamináz (ADA) cDNS-sorrendet kódoló retrovírus eredetű vektorral transzdukált CD34+ sejteket tartalmaz. Strimvelis 1-10 millió sejt/ml diszperziós infúzió GTMP (ex vivo) Feltételes MA (2016) ritka betegségek besorolás olyan adenozin- deaminázhiány okozta súlyos kombinált immunhiány szindrómás (ADA-SCID) betegek gyógyítására javasolt, akiknek nincs alkalmas emberi fehérvérsejt antigén (HLA) identikus családi őssejtdonor. 594000 € Imlygic (Amgen Europe B.V.)[24] A talimogén laherparepvek legyengített 1-es típusú Herpes simplex-vírus (HSV-1), amelyet két gén (az ICP34.5 és az ICP47) funkcionális deléciójával és egy olyan génszakasz beépítésével állítottak elő, mely emberi granulocita- makrofág kolóniastimuláló faktor sorrendet (GM-CSF-et) kódol Imlygic 106 plakk-képző egység (PFU)/ml oldatos injekció GTMP (in vivo) Standard MA (2015) nem reszekálható melanomában szenvedő felnőttek kezelésére javasolt, regionális vagy távoli metasztázisok esetén (IIIB, IIIC és IVM1a stádium), de csont, agyi, tüdő vagy egyéb zsigeri érintettség nélkül. 65000 $ Holoclar (Chiesi Farmaceutici S.p.A.)[25] A Holoclar olyan átlátszó, kör alakú lap, amelyen 300 000–1 200 000 életképes, autológ, emberi szaruhártya hámszövetsejt (79 000–316 000 sejt/cm2) található, ezeken túl átlagosan 3,5% (0,4–10%) limbalis őssejtet, valamint őssejt eredetű, átmeneti osztódó és terminálisan differenciált sejteket tartalmaz, transzportmédiumba helyezett, 2,2 cm átmérőjű fibrinrétegen. Holoclar 79 000–316 000 sejt/cm2 , szövettenyészet, élő szövet helyettesítésére TEP Feltételes MA (2016) ritka betegségek besorolás A szem fizikai vagy kémiai égése okozta, közepesen súlyos és súlyos, egyoldali vagy kétoldali limbális őssejthiányban szenvedő felnőttek kezelésére. A mintavételezéshez legalább 1–2 mm2 ép limbusszövet nélkülözhetetlen. - Provenge (Dendreon UK Ltd)[26] Autológ perifériás vérből származó, PAP-GM-CSF-fel (Sipuleucel-T) aktivált egysejtmagos sejtek. Provenge 50 x 106 CD54+ sejt/250 ml diszperziós infúzió SCTMP Visszavonva (2015-05-06) felnőtt férfiak tünetmentes vagy minimális tünetekkel járó, (nem zsigeri) áttétes, kasztráció rezisztens prosztata rák gyógyítására szolgál azoknál, akiknél a kemoterápia még nem indikált. 93000 $ MACI (Vericel Denmark ApS)[27] Porcsejt-specifikus markergéneket expresszáló, karakterizált, életképes, autológ, élőből expandált porcsejtek, CE-jelölésű, sertés eredetű, I/III. típusú, kollagén membránra ültetve. MACI 500 000 – 1 000 000 db sejt/cm2 beültetéses mátrixhoz TEP Felfüggesztett a térdporc teljes vastagságát érintő (és a módosított Outerbridge skála szerinti III. és IV. fokú), 3-20 cm2 területű, szimptomatikus károsodás helyreállítására javasolt, kifejlett csontrendszerrel rendelkező felnőtteknél. Glybera (uniQure Biopharma B.V.)[28] Az emberi lipoprotein-lipáz (LPL) gén LPLS447X variánsát tartalmazza egy vektorban. A vektor egy fehérjeburkot tartalmaz, amely az 1-es szerotípusú adenoasszociált vírusból (AAV1), a citomegalovírus (CMV) promoterből, a woodchuck hepatitis vírus egyik poszttranszkripciós regulációs eleméből és az AAV2-ből származó invertált terminális ismétlődésekből gyártott. Az alipogén tiparvovek rovarsejtek felhasználásával és rekombináns bakulovírus technológiával gyártott. Glybera 3 × 1012 genomkópia/ml oldatos injekció GTMP (in vivo) MA kivételes körülmények között (2012) Ritka betegség családi eredetű lipoprotein-lipáz hiányt (LPLD) kimutatott, és a zsírszegény diéta ellenére súlyos vagy többszöri hasnyálmirigy- gyulladásos roham alakul ki náluk. Az LPL-hiányt genetikai vizsgálattal bizonyítani kell. Az orvosi javaslat köre azokra vonatkozik, akiknek mérhető szintű LPL proteinnel rendelkezik. 1,1 millió € ChondroCelect (TiGenix N.V.)[29] Specifikus marker-fehérjéket expresszáló, élőből származó (ex vivo), szaporított, karakterizált, életképes autológ porcsejtek. ChondroCelect 10 000 sejt/mikroliter implantációs szuszpenzió TEP Visszavont kizárólag autológ alkalmazásra, és debridementtel (a defektus területének előkészítése), a defektus fedésével (biológiai membrán, lehetőleg kollagén membrán behelyezése) és rehabilitációval együtt kell felhasználni. 20000 €
Az ATMP-k európai piaca[szerkesztés]

A fejlett terápiás gyógyszerkészítmények (ATMP) jelentős eszközöket jelentenek az olyan hatékony kezelésben részesülő betegek számára, akik korlátozott vagy hiányzó terápiás lehetőségeket szenvednek. Az ATMP olyan területet jelent, ahol az európai kutatás és fejlesztés kiváló eredményeket hozott. Olaszország jelenleg vezető szerepet tölt be Európában, hiszen az európai piacon jelenleg elérhető négy közül négyet az olasz kutatók teljesen kifejlesztettek. Ezek közül az első olyan piaci engedélyezett ATMP, amely az őssejteken alapul.
Azok az ATMP-ek, amelyek sikeresen piacra dobták, csak egy része azoknak, amelyeket a klinikai vizsgálatok során fejlesztenek és gyártanak, főként egyetemi intézmények és gyakran támogatott jótékonysági szervezetek vagy állami források. Az ATMP-fejlesztés kezdetétől kezdve minden olasz termelőegység megfelelt az alkalmazandó szabályozási követelményeknek, és a rendszeresen ellenőrzés gyakorlat az illetékes hatóságoknak, mivel rendkívül fontos, hogy az ATMP-fejlesztést a szabályozási követelményeknek megfelelően hajtsák végre.[30]
A fejlett terápiás gyógyszerkészítmények (ATMP) kereskedelmi fejlesztése nagyszerű lehetőséget kínál a terápiás innováció számára, de sok kihívással szembesül a fejlesztők körében. Bár az ATMP-mező gyors ütemben halad előre, amit az elmúlt néhány évben végzett klinikai vizsgálatok egyre növekvő száma mutat, számos tényező továbbra is bonyolítja az ATMP-k bevezetését több betegségtípus gyógyító kezelésére, blokkolja a transzlációs útvonalát a kutatástól a betegig. Míg számos új kiadvány (Trounson és McDonald, 2015; Abou-El-Enein és mások 2016 a, b), valamint az innovatív gyógyszerek kezdeményezésével foglalkozó konzultáció (IMI, 2016) kiemelte az ATMP-fejlesztés főbb hiányosságait, a szabályozási és a visszatérítési kérdéseket illetően, továbbra is koherens stratégiát kell megfogalmazni ezek kezelésére azáltal, hogy az érintett érdekelt feleket egy egységes fórumba helyezzék, amelynek feladata a legsúlyosabb szűk keresztmetszetek enyhítésére irányuló delta terv kidolgozása és végrehajtása lenne.
A fejlett terápiás gyógyszerek kutatása, fejlesztése, forgalombahozatala[szerkesztés]
A gyógyszer kifejlesztése számos különálló szakaszból áll, amelyek a termék minőségének, biztonságosságának és hatásosságának bemutatására szolgálnak.
Felfedezés fázisa[szerkesztés]
Jellemzően a termékfejlesztés a pre-klinikai proof of concept (PoC) in vitro vagy in vivo betegségmodell tanulmányok elkészítését követően kezdődnek.
Nem klinikai vizsgálatok[szerkesztés]
A fejlesztendő gyógyszerjelölttel nem klinikai vizsgálatokba kezdenek a biztonságosság bizonyításához és beszerzik a betegek klinikai vizsgálatát támogató kezdeti indikáció hatásmechanizmusáról szóló jelentést.
Klinikai vizsgálatok[szerkesztés]
Ha a nem klinikai adatok alapján Klinikai Vizsgálati Engedélyt (Clinical Trial Authorisation), (CTA-alkalmazást) hagynak jóvá, a Vizsgálati Gyógyszerkészítmény (Investigational Medicinal Product) IMP-t először a Fázis I. vizsgálatban (először emberben) végzett vizsgálatoknál általános biztonságra tesztelik, ezt követően pedig a Fázis II. vizsgálatban a terápiás hatásmechanizmus (kezdeti hatékonyság) kísérletei, majd a Fázis III. vizsgálatban (pivotal, döntő) vizsgálatok a hatékonyságának megerősítésére szolgál.
- A génterápiás klinikai vizsgálatok indikációi (2016)[31]
Indikáció Daganatos betegségek Monogénes betegségek Fertőző betegségek Neurológiai betegségek Szemészeti betegségek Gyulladásos betegségek Egyéb Gén mintázat Egészséges önkéntesek Klinikai vizsgálatok száma (n) 1590 259 182 45 34 14 56 50 54 Klinikai vizsgálatok aránya (%) 64.6 10.5 7.4 1.8 1.4 0.6 2.3 2 2.2
Törzskönyvezés és forgalombahozatal[szerkesztés]
A Fázis III. klinikai vizsgálatokból származó adatokat felhasználják a forgalomba hozatali engedély iránti kérelem (marketing authorization applications (MAAs)) támogatására az EMA felé. Ha a forgalomba hozatali engedélyt (MA)-t megadják, a jóváhagyást követő jóváhagyó biztonsági adatokat a Fázis IV. klinikai vizsgálatok során meg kell szerezni az MA fenntartása érdekében.
MA utáni időszakban, a gyártási folyamat minden olyan változását, amely befolyásolhatja a gyógyszer minőségét, az EMA-nak a MA fenntartása céljából az EMA-hoz kell benyújtani ellenőrzésre és jóváhagyásra. A változtatás minőségre gyakorolt hatását a jóváhagyott termékhez képest összehasonlíthatósági tanulmányokkal kell igazolni. Szükség lehet további összehasonlító vizsgálatokra is a biztonság és a hatásosság szintjén.
A fejlett terápiás gyógyszerkészítmények gyártásának elemei[szerkesztés]
A gyógyszer gyártási folyamatának (beleértve az ATMP-t) fejlesztését úgy lehet tekinteni, hogy először a hatóanyag gyártását foglalja magában a kiindulási anyagokból és nyersanyagokból, másodszor pedig a gyógyszer gyártása a hatóanyagból és nyersanyagból és / vagy segédanyagokból.
Az ATMP előállításához szükséges kiindulási anyagok közé tartoznak az adományozott (donortól kapott) sejtek vagy szövetek, a sejtvonalak és a klónozott gének expresszált vírusvektorok (más típusú vektorok is alkalmazhatók). A nyersanyagok magukban foglalhatják a sejttenyésztő táptalajt, a szérumot és a sejt disszociációs szereket, míg a segédanyagok (a végtermék nem gyógyszerészeti összetevői) stabilizátorokat vagy krioprezervatív anyagokat tartalmaznak.
A hatóanyag, az aktív anyag /aktív gyógyszerészeti összetevő felelős a gyógyszer terápiás hatásáért, és a gyógyszerkészítmény a végső formája a készítményben lévő hatóanyagnak, amely a betegeknek történő beadásra alkalmas forma. A hatóanyagot és a gyógyszert a fejlesztési szakasz során teljes mértékben jellemezni kell a termék-specifikus minőségi jellemzők azonosításához szükséges megfelelő analitikai módszerekkel.
A kritikus minőségi jellemzők összefüggenek a biztonsággal és hatásossággal a betegekben, amelyet tipikusan a hatóanyag és / vagy a gyógyszer kibocsátási specifikációjának és stabilitási profiljának meghatározására használják. A kibocsátás-specifikációban és a stabilitási profilban (például a hatásosság, a termékhez kapcsolódó szennyezés) szereplő minőségi attribútumok meghatározása és indokolása a folyamat és a termék jellemzése során kapott adatokon alapul.
A gyártási folyamat lépéseit a folyamatparaméterek és a folyamatban lévő ellenőrzések segítségével kell ellenőrizni annak érdekében, hogy a specifikációk megvalósíthatók legyenek. Együttesen, a gyártási folyamatfejlesztéshez (inputok, folyamat lépések, analitikai módszerek, karakterizálás, kimenetek, validálás) kapcsolódó valamennyi tevékenységet CMC-nek (kémia, gyártás és ellenőrzés) nevezik.
Mesenchymális őssejtkészítmények gyártásának folyamatfejlesztése[szerkesztés]

A szomatikus sejtterápia gyógyszergyártásának a példája a folyamatfejlesztésnek. ahol az adományozott szövet kiindulási anyagából, a mesenchymális őssejteket(MSC) csontvelő aspirátumból izolálják és tenyésztéssel szaporítják. Ezek a műveletek egy köztes szakaszt jelentenek, amely egy közbenső sejt sarzst (intermediate cell batch: ICB) generál. A sejtek szaporodásával kapcsolatos valamennyi lépést (mesenchymal stem cell) MSC-specifikus IPC-ken keresztül kell ellenőrizni, például a duplázási időt, a populációs szintet, a morfológiai és a felületi markerprofil-elemzést, a sejthozamot, az életképességet és az összefolyást. Ezenkívül az ICB krioprezervációja lehetővé teszi, hogy a kiindulási anyagból elkülönített és kiterjesztett MSC-ket teljes mértékben jellemezzük annak érdekében, hogy biztosítsuk azok alkalmasságát a további termékgyártásra.
A folyamat nem folytonos jellege is mutatja lehetővé teszi a hatóanyagon (DS: drug substance) végrehajtandó teljes felszabadulási teszteket, amelyeket a végső fázisú végén lévő sejtekként definiálnak (figyelembe kell venni, hogy további ICB-ket kell bevezetni a DS előtt, ha szükséges).
Azonban, attól függően, hogy a DS-kibocsátási tesztek időzítése (a krioprezerváció előtt vagy után), valamint a DS-t (DP) generálják, a DS és a DP lényegében azonosnak tekinthető, és a DP-n csökkentett felszabadulási teszt igazolható. A DS és a DP esetében a kibocsátási előírások és a stabilitási profilok meghatározása mellett kiterjesztett jellemzést lehet fontolni a végtermékre (DP) reprezentáló sejtekre a sejtek tumorigenitási potenciáljának értékelésére a gyártási folyamat végén.[33]
A kereskedelmi siker kulcsa a folyamatfejlesztés és logisztikai szempontok[szerkesztés]
A sejtterápiás klinikai vizsgálatok sikere azon a képességen alapul, hogy életképes, hatásos terméket biztosítanak. Ez a pozitív végeredmény közvetlenül a meglévő stratégiáknak és a támogató folyamatoknak tulajdonítható. A megbízható terápiás logisztikai stratégia elengedhetetlen annak biztosításához, hogy a biológiai gyógyszerkészítmények, a terápiás anyagok és a betegek biológiai mintái életképessé váljanak a gyűjtés helyéről, a gyártáson keresztül a végső klinikai helyszínre szállításig. A sejtalapú anyagok kezelésének sikeres logisztikai stratégiájának kialakításának alapjai között néhány fontosabb és fontos tényezőt kell figyelembe venni: a megfelelő száraz-szállítási egység kiválasztása, amely egy adott hasznos teher és szállítási konfiguráció, megfelelő adatgyűjtő kiválasztása, felügyeleti lánc létrehozása, tranzitszállító értékelése és potenciális a kriogén hőmérsékleteken történő szállítás problémái.[34] Számos egyedi logisztikai kihívás jelentkezik az autológ sejtterápiák esetében – azoknál a betegeknél, ahol a beteg saját sejtjeit használják a kezelés előállításához, amelyet csak az adott betegnek adnak be. Ezzel ellentétben, az allogén sejtterápia nem kapcsolódik donorhoz, és a betegek egy releváns populációjához tartoznak. Mindezek a fenti megfontolások mindkét típusú celluláris terápiára vonatkoznak.
Az autológ terápiák két különálló sejtgyűjtést igényelnek a betegtől: tumor- és dendritikus sejtekét (aferézis). Így a végtermék átvételére, tárolására és elosztására irányuló folyamat elengedhetetlen a sikerhez. Az ellátási lánc nagyobb részletességgel történő meghatározását a folyamatábra részletezi a három magas szintű folyamattal.
Tumorsejtek gyűjtése[szerkesztés]
Logisztikai perspektívából az egész autológ sejtterápiás gyógyszergyártási folyamat a tumorsejtek gyűjtésére szolgáló készlet létrehozásával kezdődik, és a kapott mintát egy adott beteggel biztonságosan azonosítani képes. Egyedi azonosítószámot rendelünk hozzá, amelyet a gyűjtés, gyártás, terjesztés és adminisztráció egész folyamatában használunk. Az ilyen eljárások biztosítják, hogy a megfelelő termék a megfelelő betegbe kerül.
Ez a tumorsejt gyűjtő kit egy szakképzett beszállító személyzettel is rendelkezik, aki a szöveteket átmeneti tárolóhelyre szállítja. Ezzel a kittel a klinikai személyzet összegyűjti a tumormintákat és átcsomagolja egy közös raktárba szállítás céljából, ahol azt megrendelték, és beazonosítva tárolják a feldolgozásig. Ezen a ponton ismét minősített csomagolással elszállítják a gyártás helyére, ahol a gyártás megkezdéséig tárolják.
Aferézis gyűjtemény[szerkesztés]
Az időzítés és logisztika döntő fontosságú az aferézis gyűjtés szempontjából, mivel az autológ sejtterápiás gyártási folyamat kezdetén megfontolt a kapott dendritikus sejtek kézhezvételekor. A daganatgyűjtési eljárással ellentétben egyetlen átmeneti tárolási lépés sem szakítja meg a minta előrehaladását. A daganatgyűjtéshez hasonlóan azonban a folyamat egy készletből indul ki, amely magában foglalja a gyűjtéshez szükséges valamennyi összetevőt. Ez a készlet tartalmazza a páciens-specifikus azonosító címkéiket és gyűjtőtartályokat, valamint egy minősített szállító konténert. A dendritikus sejteket egy közös hordozó szállítja, egy fontos lépéssel kiegészítve: a gyártó értesítésével, hogy a szállítmány úton van. Az így kapott küldeményt a gyártó megkapja, amely megerősíti a beteg azonosságát és megkezdi a gyártási folyamatot.
Az autológ sejtterápiás gyógyszer visszatér a beteghez[szerkesztés]
A gyártás után az autológ sejtterápiás adagokat fagyasztva tárolják és betöltik egy minősített száraz, szállítási egységbe, amelyet egy közös hordozón egy elosztóhelyre szállítanak, ahol az anyagot átveszik és iktatják. Amikor ezt a sejtterápiás terméket becsomagolják, hogy elhagyja a gyártás helyét és "megszerzik" a terjesztés engedélyét, ekkor elindul egy gyógyszer a beteghez vezető útján. Az elosztó központban a személyre szóló adagokat készletbe veszik és gőzfázisú fagyasztókban tárolják, amíg a klinikai vizsgálatot végző orvos nem igényli a beteg részére. Minden kért adagot egy szakképzett személy száraz, közönséges szállító eszközzel a klinika erre kijelölt terápiás egységébe.
Standardizálás[szerkesztés]
Fontos, hogy a logisztikai stratégia kidolgozásakor minél több folyamatot és eljárást szabványosítsunk. Egyes folyamatok kívül maradnak a közvetlen ellenőrzésen. A sejtgyűjtéshez minden egyes folyamathoz utasításokat alkalmazzunk, ahol rendelkezésre áll az eljáráshoz, így a címkézés konzisztens, és (a gyűjtő KIT esetében), hogy minősített szállítási megoldást alkalmazzanak a kritikus pácienssejtek gyűjtése után történő szállításkor.
Az, hogy milyen mértékben befolyásolják a premade kitek a klinikai beavatkozás eredményét attól függ, hogy milyen mértékű a beavatkozás komplexitása. A klinikai kísérlet kimenetelét befolyásoló prematura készletek mértéke attól függ, hogy milyen eljárást alkalmaznak. A műtéti beültetést igénylő beadási készletet sokkal kritikusabban kell követni, mint egy intravénás beadással alkalmazható KIT-et. Mindenesetre azonban a cél a folyamat variáció csökkentése és biztosítsa, hogy a klinikai eredmények tükrözzék az adott terápia hatékonyságát, nem pedig kezelési változásait.
Csomagolás és szállítás minősítése[szerkesztés]
Az anyag sikeres szállítása minden logisztikai stratégia elsődleges célja. Vegyük figyelembe, hogy a bemutatott egyszerű áramlási diagram öt különböző szállítmány három különböző anyagból négy különböző hőmérsékleten történik:
- A beteg daganatszövetér (vagy más alkalmazandó szövetet) összegyűjtik és ellenőrzött környezeti hőmérsékleten szállítanak egy tárolási helyre -80 °C-on tartósítva.
- Szükség esetén a beteg szövetét szárazjégen szállítják a sejtterápiás gyártóberendezéshez.
- Ugyanaz a beteg aferézis gyűjtött mintái hűtéssel kerülnek (2-8 °C) közvetlenül a gyártó létesítménybe.
- A kész terápiás adagokat ezután ömlesztve szállítják a gyártó létesítmény tároló / elosztó helyre, száraz szállítótartályok alkalmazásával, kriogén hőmérsékleten (Jellemzően -40 °C alatti hőmérséklet).
- Ezután a kész sejtterápiás adagokat továbbítják (ismét száraz szállítási egységekben) a betegek beadásának klinikai helyére.
Az öt tranzitpont mindegyikénél kritikus fontosságú, hogy olyan minősített szállítási megoldásokat használjanak, amelyeket úgy terveztek, hogy kielégítsék az egyes hasznos teherfajták sajátos követelményeit a hőmérséklet, az időtartam, a környezetvédelem és a szállítás során felmerülő szélsőségek kezelésére. Ezeket a specifikációkat nem szabad összetéveszteni a szállítási megoldások gyártó által gyakran kiadott durva minősítésekkel. Ezek az információk hasznosak lehetnek a lehetséges tesztelési megoldások kiválasztásához.
Bár nem tartoznak a szigorú tesztelésekhez, amely annak biztosításához szükséges a szállítandó anyag tökéletes állapotban célba érjenek. A minősített szállítási megoldás kritikus fontosságú annak biztosítására, hogy a sejtterápia a klinikai hatékonyságon alapuljon, nem pedig a csomagolási vagy szállítási hibák által okozott hiányosságok alapján. Az ábra kiemeli azokat a folyamatpontokat, amelyeken a szállítás és a csomagolási minősítésre van szükség.
EU szabályozás[szerkesztés]

Az EU-ban az 1394/2007 / EK rendelet 2008 óta hatályban van, és az ATMP-ekre vonatkozó közös szabályozási keret.[35] A rendelet fő előnyei közül az automatikus hozzáférést biztosít az ATMP-kre vonatkozó uniós engedélyezés központi útjához, azzal a céllal, hogy javuljon ezek elérhetősége minden tagállamban a betegek számára. A rendelet létrehozta a Fejlett Terápiák Bizottságát (CAT) is, amely) az EU egész területén releváns szakértelmet bonyolít le, és többek között az ATMP-k korai fejlesztését támogató osztályozási és tanúsítási eljárásokat is létrehozta.
EMA szerepe[szerkesztés]
Az engedélyezési követelmények figyelembe vétele elsőrendű elvárás, hogy az ATMP gyártás egyedülálló sajátosságokat feltételez (decentralizáció, hospitalizáció). Például az ATMP gyártását engedélyezzék különböző földrajzi helyeken (decentralizáció), és hogy bizonyos termékek esetében a termelés későbbi szakaszaiban az ágy melletti helyre kell kerülnie a gyártástechnológiának. Ennek az az oka, hogy az érintett területeknek – a kórházak is – rendelkezniük kell gyártási engedéllyel. A nemzeti szintű harmonizáció hiányát is rendezni kell, mivel egyes tagállamok lehetővé teszik a kórházak számára, hogy gyártási engedélyt szerezzenek, míg mások nem. Az érdekeltek ezért több információt és harmonizációt kértek.
- Az EMA belső szabályozási folyamatainak egyszerűsítése az ATMP-k számára
- A korai hozzáférési eszközök (PRIME, adaptív utak, ITF, tudományos tanácsadás, tanúsítás) használatának elősegítése és HTA párhuzamos tanácsok
- A tanúsítások nem kkv-k számára történő megnyitását, és értékük megerősítése
- A nemzeti előírások közzététele (GMO-k és szövetek, sejtek és vérkészítmények vonatkozásában) és elmozdulás a nagyobb egységesség felé
- Alkalmazási dokumentumok összehangolása (pl. CTA-k és tudományos tanácsadási kérelmek)
- A globális követelmények összehangolása (az Egyesült Államokkal és Japánnal az ICH-n keresztül)
- Iránymutatások a vizsgálati gyógyszerekről és az összehasonlíthatóságukról
- A ritka betegségek ATMP-khez való alkalmazkodása
- A betegadatok nyilvántartásának előmozdítása a hatékonyságra és biztonságra vonatkozó strukturált adatok összegyűjtésével (a adatbázisok harmonizálása, az elektronikus egészségügyi nyilvántartás használatának elősegítése)
Gyártástechnológia harmonizáció[szerkesztés]
A javasolt megoldások közé tartozik az innovatív technológiák (pl. zárt rendszerek) és gyártási modellek (pl. decentralizált gyártás), nagyobb rugalmasság bevezetése. A szabályozók ugyancsak elősegíthetik a gyártási helyszínek fejlesztését, szolgáltatásként, felhasználásával a kis- és középvállalkozások (KKV) és egyéb finanszírozás, így több lehetőség álljon rendelkezésre különböző ATMP-eket fejlesztői felé. Ezek a javaslatok időszerűnek tekinthetők, mivel az Európai Bizottság jelenleg felülvizsgálja az iránymutatás a GMP követelményeivel szemben az ATMP-k számára. konzultációt indít.
Az iparág üdvözölné a segédanyagokra vonatkozó specifikus útmutatást és a forgalomba hozatallal kapcsolatos master fájlrendszert a segédanyagokra és a nyersanyagokra vonatkozó engedélyezési célok tekintetében. A nem klinikai követelmények kezelésében fontolóra kell venni az új fejlesztési eszközöket is (pl. organoidok, extrapoláció, modellezés / szimuláció, biomarkerek stb.).
Géntechnológiával módosított szervezetek (GMO-k)[szerkesztés]
A GMO irányelv (2001/18 / EK irányelv) nem kifejezetten a gyógyszerek és gyógyszerkészítmények tekintetében készült. A hiányosságokat e tekintetben a tagállamokban végrehajtott eltérések komplikálják. Mivel a követelmények eltérnek a tagállamok között, a GMO-vizsgálatok integrálása a klinikai vizsgálatok során az engedélyezés kihívást jelent, különösen a multicentrikus klinikai vizsgálatok keretében.
A fejlesztők nagyobb egységességre szólítottak fel, és kezdeti lépésként egy központi adatbázis létrehozását javasolta angol nyelven, amely felsorolja a GMO-értékelés követelményeit és határait minden tagállamban. GMO értékelést szorosabbá kell tenni a klinikai vizsgálati engedélyhez (CTA) kötve, és időbeli ütemezéssel kell rendelkeznie a GMO-knak a CTA-khoz igazodó értékelésben. Ezen kívül harmonizált alkalmazásokra van szükség a klinikai vizsgálati portál és adatbázis felhasználásával a tervezett folyamatokhoz, hasonlóan az etikái bizottsághoz. Néhány érdekelt fél maga is felszólított a GMO-irányelv módosítására.
Sejt- és szövetterápiás termékek[szerkesztés]
Egy másik olyan terület, amely a további harmonizációra vár. A kiindulásként használt sejtekre és szövetekre vonatkozik az ATMP-k előállításával kapcsolatban szükséges változtatás. Az érdekeltek számára egyszerűbbé tétele a szövetekről és a sejtekről szóló irányelv (2004/23 / EK irányelv) végrehajtásával kapcsolatos releváns jogszabályok tekintetében. Kiindulópontként a harmonizációért felelősök létrehozhattak egy portált, amely az EU sejt- és szövetbiztonsági hatóságainak tájékoztatást nyújtanak a sejt- és szövet kiindulási anyagok országos tesztelésére vonatkozó további követelményekről. A további harmonizációnak elő kell segítenie a kiindulási anyagok mozgását a (EU-n belül és kívül), és csökkenti a sejtek újbóli tesztelésének terhét a szövetei ATMP-k gyártásának megkezdése előtt. A tesztelés ezután a szempontokra összpontosíthat amelyek az ATMP-k jellegére vonatkozóan.

Fejlett Terápiás Gyógyszerkészítmények Bizottsága (CAT: Committee for Advanced Therapies)[szerkesztés]

Európai Gyógyszerhatóság (EMA) humán alkalmazásra szánt orvosi termékekkel foglalkozó bizottságának (CHMP) véleményező szerve a fejlett terápiás készítmények értékelésével foglalkozó szakértői bizottság (CAT, Committee for Advanced Therapies), amely részletes értékelő jelentéseket készít a CHMP részére.[36] Annak érdekében, hogy az ATMP összetett és új termékek értékeléséhez a rendelkezésre álló legjobb szakértelmet alkalmazzák, az ATMP-rendelet létrehozta az EMA, a fejlett terápiákkal foglalkozó bizottságot (CAT) (az ATMP-rendelet 7. fejezete).
Tagjai[szerkesztés]
A CAT egy multidiszciplináris csoport, amely mintegy 37 tagból áll, széles körű szakértelemmel (beleértve a határokon átnyúló tudományágakat, például az orvostechnikai eszközöket):
- az emberi felhasználásra szánt gyógyszerek bizottságának (CHMP) 5 tagja (plusz helyettesei);
- minden egyes EU-tagállamban 1 tag (plusz póttag), amelyet még nem képvisel az 5 CHMP-tag;
- 2 tag (plusz póttagok), akik a Bizottság által kijelölt orvosokat képviselik;
- 2 tag (plusz póttag), akik a Bizottság által kijelölt betegegyesületeket képviselik.
Fontos megjegyezni, hogy az összes CAT-tagot speciális tudományos képesítéssel és szakértelemmel választják ki a fejlett terápiákhoz kapcsolódó területeken, beleértve az orvostechnikai eszközöket, a szövetkezelést, a gén- és sejtterápiát, a biotechnológiát, a műtétet, a farmakovigilanciát, a kockázatkezelést és az etikát. Ezenkívül további szakértők is konzultálhatnak a bizottság által szükségesnek ítélt esetekben. A CAT tagokat 3 évre megújítható időtartamra nevezik ki, és ugyanazon időtartamra megválasztanak egy bizottsági elnököt.
| „ | A fejlett terápiás gyógyszerkészítmények értékelése gyakran nagyon sajátos szakértelmet igényel, ami túlmegy a hagyományos gyógyszerészet területén és más ágazatokkal – mint például a biotechnológia és az orvostechnikai eszközök – szomszédos területeket is lefed. Emiatt helyénvaló az ügynökségen belül a fejlett terápiákkal foglalkozó bizottság létrehozása, amely felelős az egyes fejlett terápiás gyógyszerkészítmények minőségéről, biztonságosságáról és hatásosságáról szóló véleménytervezet elkészítéséért, amelyet az ügynökség emberi felhasználásra szánt gyógyszerek bizottsága hagy véglegesen jóvá. A fejlett terápiákkal foglalkozó bizottsággal konzultációt kell folytatni továbbá minden egyéb olyan gyógyszerkészítmény értékelése tekintetében, amely annak illetékességi területébe tartozó különleges szakértői vizsgálatot igényel.” | ” |
| – Az Európai Parlament és a Tanács 1394/2007/EK rendelete ( 2007. november 13.) a fejlett terápiás gyógyszerkészítményekről[37] | ||
Fokozottan hangsúlyos terület az őssejtek terápiás alkalmazása a klinikumban. Texas-ban több, mint 20, az USA‐ban 100‐nál több őssejt klinika, több millió dolláros jövedelmekkel, az FDA engedélye nélkül gyógyítanak. Az EMA-CAT állásfoglalása az “őssejt-turizmus” nevű jelenség kapcsán, a nem igazolt terápiákkal a betegek pénzén végeznek “ember-kísérleteket”!
Hatásköre[szerkesztés]
A Fejlett Terápiás Bizottság (CAT) központi szerepet játszik a fejlett terápiás gyógyszerek tudományos értékelésében . Ez biztosítja a fejlett terápiás gyógyszerek értékeléséhez szükséges szakértelmet.
Az értékelési eljárás során a CAT véleménytervezetet készít a fejlett terápiás gyógyszer minőségéről, biztonságáról és hatásosságáról. Ezt elküldi az emberi felhasználásra szánt gyógyszerek bizottságának (CHMP). A CAT- vélemény alapján a CHMP véleményt fogad el, amely a gyógyszert az Európai Bizottság engedélyezi vagy nem engedélyezi. Az Európai Bizottság végleges döntést hoz a CHMP véleménye alapján.
- ajánlásokat tesz a fejlett terápiás gyógyszerek osztályozására vonatkozóan ;
- értékeli a kkv-k számára a minőségi és nem klinikai adatok igazolására irányuló kérelmeket , amelyek után az Ügynökség tanúsítványt állít ki;
- hozzájárul a tudományos tanácsadáshoz a fejlett terápiás gyógyszerek területén;
- részt vesz minden olyan eljárásban, amely a vállalkozások számára nyújt tanácsot az ATMP-k hatékonysági monitoringjáról, farmakovigilanciai és kockázatkezelési rendszereiről ;
- a CHMP kérésére minden olyan gyógyszerre vonatkozóan tanácsot ad, amely a minőségének, biztonságosságának vagy hatásosságának értékeléséhez szükségessé teheti az ATMP-kben szerzett szakértelmet;
- tudományosan segíti az 1394/2007 / EK rendelet célkitűzéseinek teljesítésével kapcsolatos dokumentumok kidolgozását;
- hozzájárul a fejlett terápiás gyógyszerek fejlesztését ösztönző környezethez;
- az Európai Bizottság kérésére tudományos szakértelmet és tanácsadást nyújt az innovatív gyógyszerek és terápiák fejlesztésével kapcsolatos kezdeményezésekhez .
USA szabályozás[szerkesztés]
A Center for Biologics Evaluation and Research (CBER) szabályozza a sejtterápiás termékeket, az emberi génterápiás termékeket és bizonyos sejt- és génterápiával kapcsolatos eszközöket. A CBER a közegészségügyi szolgálatról szóló törvényt és a szövetségi élelmiszer- és kozmetikai törvényt is felhasználja, mivel lehetővé teszi a felügyelet alapszabályát. A sejtterápiás termékek közé tartoznak a celluláris immunterápiák, daganatellenes vakcinák és más típusú autológ és allogén sejtek bizonyos terápiás indikációkban, beleértve a hematopoetikus őssejteket és a felnőtt és embrionális őssejteket.
Az emberi génterápia egy genetikai anyag alkalmazása géntermék expressziójának módosítására vagy manipulálására vagy az élő sejtek biológiai tulajdonságainak megváltoztatására terápiás célra. A CBER mind a celluláris, mind a génterápiás készítményeket jóváhagyta.
- Jóváhagyott sejt- és génterápiás termékek az Office of Tissues és Advanced Therapies (OTAT) licencelt termékeit:
- ALLOCORD (HPC Cord Blood) SSM Glennon bíboros Gyermek Gyógyászati Központ
- LAVIV (Azficel-T) Fibrocell Technologies
- MACI (autológ tenyésztett kondrociták sertés-kollagén membránon) Vericel Corp.
- CLEVECORD (HPC Cord Blood) Cleveland Cord Blood Center
- GINTUIT (allogén kulturált keratinociták és fibroblasztok szarvasmarha kollagénben) Organogenesis Incorporated
- HEMACORD (HPC, köldökvér) New York Blood Center
- Ducord, HPC Cord Blood Duke Egyetem Orvostudományi Kar
- HPC, Cord Blood Clinimmune Labs, Colorado University of Cord Blood Bank
- HPC, Cord Blood – LifeSouth Közösségi Vér Centrumok, Inc.
- HPC, Cord Blood – Bloodworks Bloodworks
- IMLYGIC (talimogene laherparepvec) BioVex, Inc., az Amgen Inc. leányvállalata
- KYMRIAH (tisagenlecleucel) Novartis Pharmaceuticals Corporation
- LUXTURNA Spark Therapeutics, Inc.
- PROVENGE (sipuleucel-T) Dendreon Corp.
- Steril vértestvérgyűjtő egység antikoaguláns citrát-foszfát-dextrózoldattal USP (CPD) MacoProductions, SAS
- YESCARTA (axicabtagene ciloleucel) Kite Pharma, Incorporated
Az Egyesült Államokban a celluláris és a génterápiával kapcsolatos kutatások és fejlesztések gyors ütemben nőnek, számos termék pedig továbbfejleszti a klinikai fejlődést. A klinikai vizsgálatok szabályozási felügyeletén túlmenően a CBER proaktív tudományos és szabályozási tanácsokat nyújt az orvosi kutatóknak és a gyártóknak az új termékfejlesztés területén.[38]
Kutatás és fejlesztés[szerkesztés]
A fejlett terápiás gyógyszerkészítmények (ATMP), amelyek génterápiás készítményeket, szöveti biomérnöki termékeket és szomatikus sejtterápiákat tartalmaznak, számos olyan állapot kezelésére képesek úgy, hogy újraformálják a terápiás eljárásokat, különösen olyan betegségek területén, ahol a hagyományos eljárások nem megfelelőek. Az ATMP-kezelések potenciális jelöltjei közé tartoznak a súlyos, nem kezelhető vagy krónikus betegségek, és számos klinikai vizsgálat jelenleg folyamatban van olyan betegségekben, mint a rák, a szív- és érrendszeri betegségek, az izomrendszer és az immunrendszer rendellenességeiben.[39] Ha az ATMP-k teljesítik ígéretüket a betegek innovatív kezelésében, a szabályozóknak olyan szabályozási környezetet kell segíteniük, amely ösztönzi az innovációt és a biztonságot az egészségügyben, és végső soron megkönnyíti a betegeknek az új terápiákhoz való hozzáférését.
A fejlett terápiás gyógyszerkészítmények (ATMP) kutatása és fejlesztése Európa-szerte és világszerte aktív az utóbbi években. Az engedélyezett termékek száma azonban továbbra is alacsony. A közeljövőben az Európai Unió (EU) legfontosabb várható változása a klinikai vizsgálatokról szóló rendeletre (536/2014) vonatkozik, amely 2018-ban lép hatályba. Ezzel az új keretrendszerrel harmonizál és gyorsabbak a várható klinikai vizsgálatok, amelyek várhatóan támogatják az új innovációk bejutását az EU piacára. Az EU-ban 2010-2015-ben végzett klinikai vizsgálatok során az ATMP-ekről készült felmérést az ATMP-fejlesztések trendjeinek 2012-ben közzétett korábbi felmérése óta folytatják az EU-ban. Az eredmények szerint az ATMP-kkel végzett klinikai vizsgálatok száma lassan növekszik az EU-ban. A hangsúly még mindig a korai fejlesztés, és a projekteket elsősorban a kis és középvállalkozások, az egyetemi klinikák és a kórházak végzik.
Az onkológia a klinikai fejlődés fő területe. Azonban a sejt-alapú termékek és génterápiás gyógyszerek közötti egyensúly ezen a területen a jövőben változhat az új T-sejtes technológiák miatt. Számos korlát és kihívás van azonosítva az ATMP fejlesztéséhez, amely arányos szabályozási követelményeket igényel. Másrészt egy ilyen új terület esetében a fejlesztőknek aktívaknak kell lenniük a lehetséges korlátok mérlegelésekor, és aktívan együtt kell működniük a hatóságokkal, hogy megoldásokat kereshessenek.
Fejlesztés szakasza[szerkesztés]
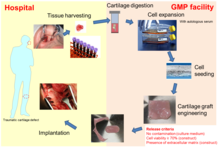
Az ATMP-k lényegében rendkívül bonyolult, komplex összetevőkből álló termékek, amelyek különböző biológiai anyagokból származnak, például sejtekből, szövetekből vagy vírusvektorokból, és egyedi jellemzőik különleges megközelítést igényelnek a kutatásban és a fejlesztésben.[40]
A fejlesztés szakaszában a fejlesztő feladata a sejt kiindulási anyag homogenitásának biztosítása és a folyamatos nyersanyagellátás biztosítása; ezen túlmenően bizonyos gyártási követelmények nem feltétlenül alkalmazhatók minden ATMP-hez, és kihívások vannak olyan kérdésekkel kapcsolatban, mint például a kiforratlan termelési technológiák fejlesztése a kereskedelmi gyártáshoz, a folyamat validálásához és a termék jellemzéséhez való korszerűsítés összetettsége.
Ezen túlmenően az ATMP-k fejlesztői, akik gyakran kisvállalkozások vagy spin-offok (inkubátorok) az akadémiákból, támogatást igényelnek a szabályozási keretbe való navigáláshoz, és részesüljenek a tőkebefektetések és az ösztönzők további hozzáférhetőségéből.
Míg az ipar és más érdekeltek elismerik a magas színvonalú igényt, kérték hogy a korai fejlesztés szakaszaiban rugalmasabbá tegyék a követelményeket. Konkrét javaslatok a sejt alapú termékek esetében az alacsony kockázatú (nem lényegileg manipulált) mivel ezek a transzplantációk és az ATMP-k közötti határvonalnak tekinthetők. Hasonlóképpen pragmatikusabb megközelítést kell alkalmazni a folyamat validálási követelményeinek kezelésére számos ATMP-t, mivel nehéz a szabványos gyógyszerek tételek számának előállítása.
A fejlesztendő termékek haszon/ kockázat egyensúlyának kérdésében megjegyzendő, hogy a jelenlegi gyakorlat főként a kockázatokra összpontosít, és további hangsúlyt helyezett a várakozásokra, ami reális előnyökkel jár, különösen akkor, ha a betegek gyógyíthatatlan betegségek és megfelelő kezelésekkel nem rendelkeznek. Azt is figyelembe veendő, hogy a jelenlegi kockázat alapú megközelítés nyomást gyakorol a Minősített Személyekre (QP), ugyanis a különböző tagállamokban található QP-k eltérő értelmezések szerint engedélyezhetik a termékek gyártását. Az érdekelők a kockázatalapú megközelítés felülvizsgálatát szükségesnek látják és további útmutatást kérnek a QPS-hez.
A haszon-kockázat egyensúlyának gondos mérlegelése a korai fejlesztési stratégia részét kell képeznie, és a szabályozó hatóságoknak (köztük a HTA-kkal) foglalkozniuk kell a folyamat korai szakaszában annak érdekében, hogy a követelményeket ennek megfelelően ki lehessen igazítani. Informális párbeszéd például biztonságos kikötőkkel, például az Innovation Task Force (ITF) hálózattal formálisabb viták révén. Tudományos tanácsra van szükség. A CAT-val együttműködve a tudományos tanácsadás jó eszköz a korai párbeszédhez, és az ellenőrök, a HTA-k és a megbízók bevonásával javítható. Ugyanakkor nem minden kkv-nak vagy akadémiai vállalkozásnak van erőforrása a tudományos tanácsadáshoz.
Jegyzetek[szerkesztés]
- ↑ Movie S1 from Feliciano D, Zhang S, Nasrallah C, Lisgo S, Bordey A (2014). "Embryonic Cerebrospinal Fluid Nanovesicles Carry Evolutionarily Conserved Molecules and Promote Neural Stem Cell Amplification". PLOS ONE. DOI:10.1371/journal.pone.0088810. PMID 24533152. PMC: 3923048
- ↑ A géneken és sejteken alapuló új gyógyszerkészítményekre vonatkozó uniós szabályok
- ↑ http://p-bio.org/wp-content/uploads/2016/06/ATMP-Handbook-Drug-Development-and-Regulation-Bioreg-Project.pdf
- ↑ Archivált másolat. [2018. január 10-i dátummal az eredetiből archiválva]. (Hozzáférés: 2018. január 10.)
- ↑ Archivált másolat. [2018. január 9-i dátummal az eredetiből archiválva]. (Hozzáférés: 2018. január 22.)
- ↑ Insight Series Briefing – Advanced Therapeutics 12 © Defined Health, 2018
- ↑ Humán blasztocisztákból származó embrionális őssejtvonalak. Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, Jones JM Tudomány. 1998 nov. 6; 282 (5391): 1145-7.
- ↑ Etikus határmunka az embrionális őssejt-laboratóriumban. Wainwright SP, Williams C, Michael M, Farsides B, Cribb A Sociol Health Illn. 2006 szept. 28 (6): 732-48
- ↑ PEI. (2016). Fejlett terápiás gyógyszerek (ATMP) . Elérhető a következő címen : http://www.pei.de/EN/medicinal-products/advanced-therapy-medicinal-products-atmp/advanced-therapy-medicinal-products-atmp-node.html Archiválva 2018. január 23-i dátummal a Wayback Machine-ben
- ↑ Forrás: Európai Gyógyszerügynökség.
- ↑ Legal wording of key definitions, according to Article 2.1 Regulation (EC) No 1394/2007
- ↑ http://www.ogyei.gov.hu/dynamic/gyogyszereink_2016_2.pdf
- ↑ Russo E. (2005) Follow the Money—The Politics of Embryonic Stem Cell Research. PLoS Biol 3(7): e234. doi:10.1371/journal.pbio.0030234
- ↑ Alliance for Regenerative Medicine. Annual Data Report on gene and cellular therapies and the regenerative medicine sector. (2016).
- ↑ The CAR T-Cell Race | The Scientist Magazine. Available at: http://www.the-scientist.com/?articles.view/articleNo/42462/title/The-CAR-T-Cell-Race/. (Accessed: 18th April 2017)
- ↑ Bersenev, A. Total number of CAR cell therapy trials listed in databases. Figshare. (2017). doi:10.6084/m9.figshare.4628557
- ↑ Szövetség a regeneratív orvostudományi adatjelentéshez 2016.
- ↑ Search on www.clinicaltrials.gov, accessed 08/05/2017.
- ↑ Boeckh, M. Current antiviral strategies for controlling cytomegalovirus in hematopoietic stem cell transplant recipients: prevention and therapy. Transpl. Infect. Dis. 1, 165–78 (1999). . Ljungman, P., Hakki, M. & Boeckh, M. Cytomegalovirus in hematopoietic stem cell transplant recipients. Hematol. Oncol. Clin. North Am. 25, 151–69 (2011).
- ↑ Riley, J. L., June, C. H. & Blazar, B. R. Human T Regulatory Cell Therapy: Take a Billion or So and Call Me in the Morning. Immunity 30, 656–665 (2009).
- ↑ Cell & Gene Therapy Insights - ISSN 2059-7800
- ↑ Union Register of not active medicinal products for human use > Zalmoxis
- ↑ Union Register of medicinal products for human use > Strimvelis
- ↑ Union Register of medicinal products for human use > Imlygic
- ↑ Public Health - Union Register of medicinal products > Holoclar
- ↑ Union Register of not active medicinal products for human use > Provenge
- ↑ Union Register of not active medicinal products for human use > MACI
- ↑ Union Register of not active medicinal products for human use > Glybera
- ↑ Union Register of not active medicinal products for human use > ChondroCelect
- ↑ 10 Years Experience With Advanced Therapy Medicinal Products: Past, Present and Future November 10th, 2017 Istituto Superiore di Sanità Aula Pocchiari – Roma - Italy
- ↑ The Journal of gene Medicine 2017. John Wiley & Sons Ltd.[halott link]
- ↑ a ong-Jun Dai, Mani R. Moniri, Zhi-Rong Zeng, Jeff X. Zhou, Jarrett Rayat, Garth L. Warnock által módosított formában. "A mezenhimális őssejtek potenciális következményei a rákterápiában". Cancer Letters, Volume 305, Issue 1, June 1, 2011, Pages 8-20.
- ↑ European Medicines Agency. EMEA/ CHMP/410869/2006 – Draft Guideline on human cell-based medicinal products. 2007. [2018. június 15-i dátummal az eredetiből archiválva]. (Hozzáférés: 2021. március 5.)
- ↑ O’Donnell D. Commercially Successful Cell Therapies: Navigating The Ultra Cold Chain Distribution Minefield. Fisher BioServices Blog 17 June 2013; http://blog. fisherbioservices.com/bid/304873/ Commercially Successful Cell Therapies - Navigating the Ultra Cold Chain Distribution Minefield
- ↑ Regulation (EC) no 1394/2007 of the European Parliament and of the Council of 13 November 2007 on Advanced Therapy Medicinal Products and amending Directive 2001/83/EC and Regulation (EC) No 726/2004.
- ↑ Javaslat: az Európai parlament és a Tanács rendelete a fejlett terápiás gyógyszerkészítményekről, valamint a 2001/83/EK irányelv és a 726/2004/EK rendelet módosításáról
- ↑ Az Európai Parlament és a Tanács 1394/2007/EK rendelete ( 2007. november 13.) a fejlett terápiás gyógyszerkészítményekről, (10) preambulumbekezdés
- ↑ https://www.fda.gov/BiologicsBloodVaccines/CellularGeneTherapyProducts/
- ↑ Hanna E, Remuzat C, Auquier P, et al. Advanced therapy medicinal products: current and future perspectives. J Mark Access Health Policy 2016;
- ↑ Ardigò D, Toschi M. Challenges and opportunities for successful development and approval of ATMPs. Regulatory Rapporteur 2016.
Források[szerkesztés]
- https://www.gov.uk/guidance/advanced-therapy-medicinal-products-regulation-and-licensing
- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4846788/
- https://web.archive.org/web/20120912042722/https://www.ich.org/fileadmin/Public_Web_Site/Training/ASEAN_Q5C_workshop_May_2011/SESSION_IVb_ATMPs.pdf
- http://www.ogyei.gov.hu/dynamic/gyogyszereink_2016_2.pdf
- http://www.hbcs.hu/uploads/jogszabaly/331/fajlok/20_2010_eumr.pdf
- [[https://web.archive.org/web/*/http://epa.niif.hu/00800/00878/01295/pdf/L_2007_324_0121_0137.pdf halott link] http://epa.niif.hu/00800/00878/01295/pdf/L_2007_324_0121_0137.pdf]
- https://www.cordbloodcenter.hu/az-ujszulottektol-levett-ossejtek-egyre-novekvo-hasznalata/
További információk[szerkesztés]
- https://web.archive.org/web/20161113072930/http://www.cellforcure.com/atmp-regulation/
- https://web.archive.org/web/20140210210428/http://www.wil-zone.hu/szakmaianyagok/GMP_jog_v%C3%A1ltoz%C3%A1s_2102-2013_13.pdf
- Molekuláris terápiák, szerk.: Balajthy Zoltán Írták: Aradi János, Balajthy Zoltán, Csősz Éva, Scholtz Beáta, Szatmári István, Tőzsér József, Varga Tamás „Az orvosi biotechnológiai mesterképzés megfeleltetése az Európai Unió új társadalmi kihívásainak a Pécsi Tudományegyetemen és a Debreceni Egyetemen” Azonosítószám: TÁMOP-4.1.2-08/1/A-2009-0011
- Investment for Advanced Therapies Report